Son-of-a-fish....
I have to show this one to you guys. Despite some great new advances recently, we really still do lag behind a lot of other biological fields when it comes to statistics. It is getting better all the time, especially with new bioinformaticians coming from other fields into ours. We'll have it all down soon and I'm excited for all the advances that keep coming.
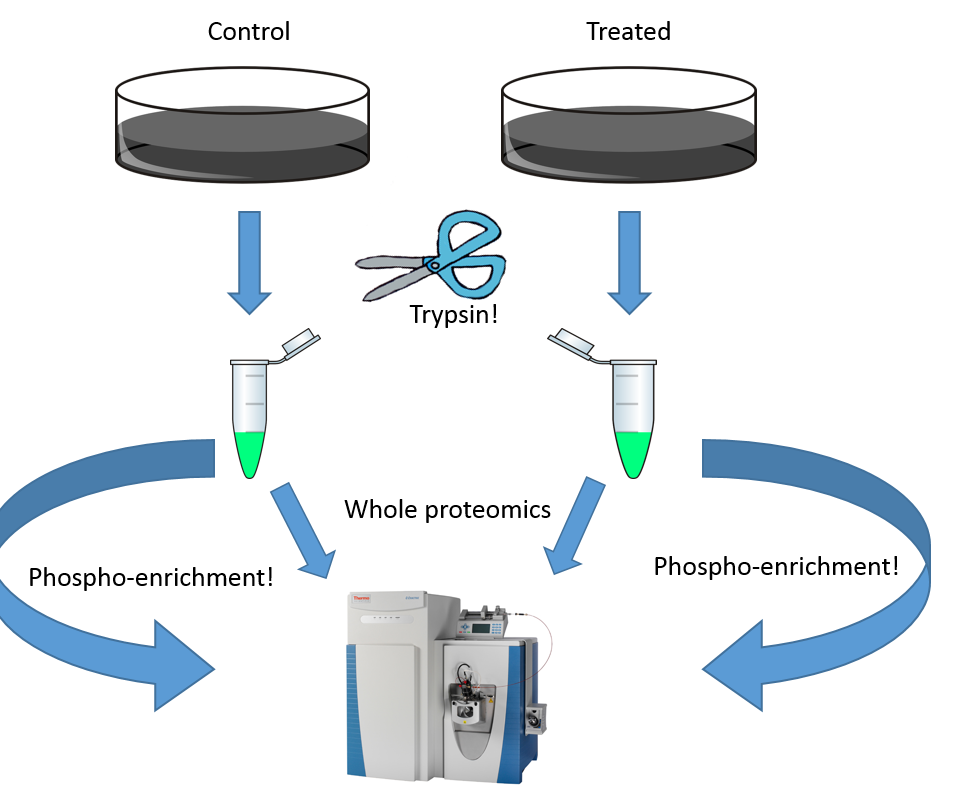
In terms of bioinformatics, we have two studies out there that have some pretty big targets painted on them. These studies are our first 2 drafts of the human proteomes. They are awesome studies of remarkably high level sample prep, instrumentation and methodologies. However, both relied heavily on the statistics tools that we all use in our daily work. In data sets this large and complex, our typical tools, namely the 1% false discovery rate, seems particularly weak. 1% bad matches sounds pretty good until you have a billion observations.
The title of this paper is a good one: "Solution to Statistical Challenges in Proteomics Is More Statistics,Not Less." and really highlights the value of more, and increasingly robust, statistical analyses in our proteomics studies when we are looking at increasingly larger datasets.
It makes me sad (and I find it just a little scary) because I don't personally have the background to effectively evaluate the quality of the statistics. Now that we're in this era where our instruments generate more data than we could ever possibly manually examine we are going to need to rely more and more on these algorithms to sort things out and we're going to have to find a way to let go of a little control and trust them.
This paper is a good, short, open access(!) read.