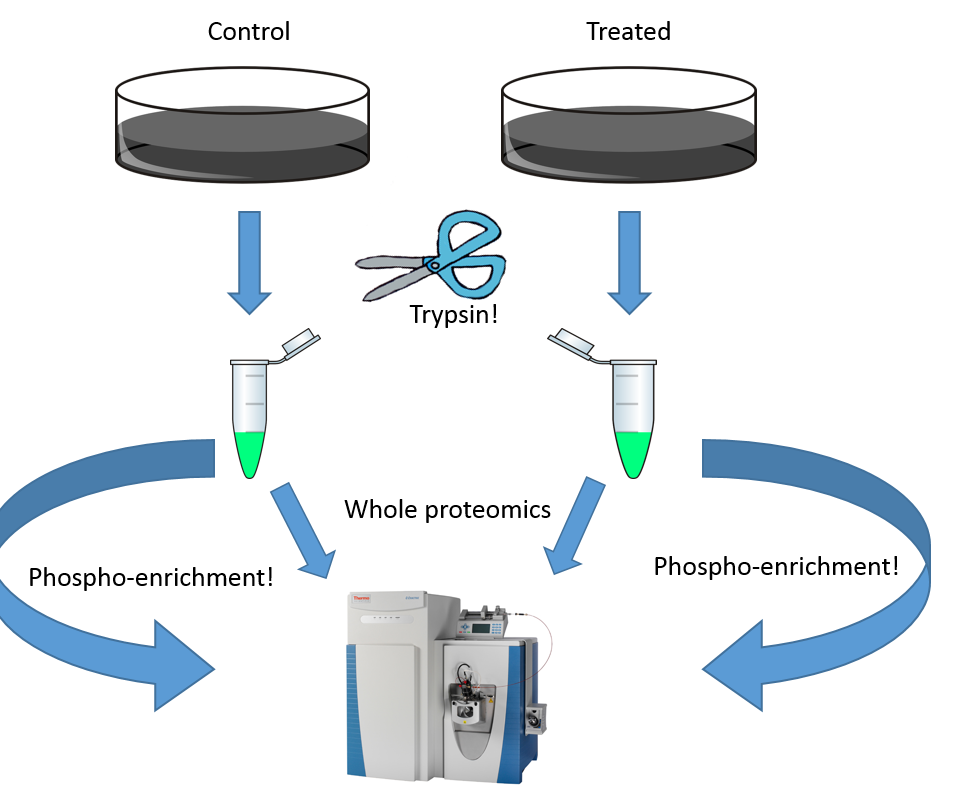
Aging sucks. You spend all this time looking forward to it when you're a puppy. Looking toward the day when you can drive and stay out all night dancing and are trusted to go outside and do your business and come back inside when you're done. But then it should stop. And progress is being made here and there on that front, but before we can stop it we need to understand it a little better. There are tons of good papers out there studying aging in mice and worms and zebra fish. Its tougher to do this stuff with human beings.
For an interesting model of how we could analyze human aging check this paper from Christina Menni et al.,(open access!). It isn't my typical proteomics paper but there is a lot of interesting stuff here.
First of all, they have a big group of human female twins, called the TwinsUK cohort. Twins are great for studying all sorts of things, but are most often called upon for nature vs. nurture type studies. Another awesome use of twins? Biological replicates! What they did here was take plasma from this large group of British ladies and performed standardized RNA-Seq analysis looking for changes that could be aging related.
Another great thing about studies of this kind is that once you get a bunch of human volunteers lots of researchers want to test lots of different technologies on their samples. Turns out that most of these volunteers had already had plasma drawn and ran through a kind of protein array technology called SOMAScan (you can read about it here if you are interested!)
What did they find? Clear, circulating biomarkers that are detectable at the RNA level and carry over to the protein. When you go for overlaps of technologies between RNA and protein you are going to typically see a pretty small list, and this study is no exception, but they found a cool little list.